Biologic therapeutics such as monoclonal antibodies and biosimilars are complex proteins that are susceptible to post-translational modifications (PTMs). These chemical modifications can affect the performance and activity of the biologic, potentially resulting in decreased potency and increased immunogenicity. Such modifications include glycosylation, deamidation, oxidation and disulfide bond shuffling. These PTMs can be signs of protein degradation, manufacturing issues or improper storage. Several of these modifications are well characterized, and methods exist for detecting them during biologic manufacture. However, disulfide shuffling is not particularly well characterized for biologics, and no methods exist to easily detect and quantify disulfide bond shuffling in biologics.
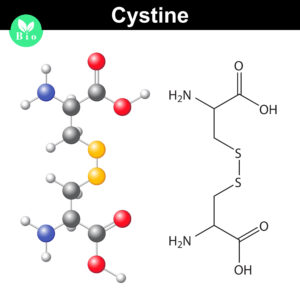
Normally the cysteines in a protein will pair with a predictable or “normal” partner residue either within a polypeptide chain or between two polypeptide chains when they form disulfide bonds. These normal disulfide bonds are important for final protein conformation and stability. Indeed, disulfide bonds are considered an important quality indicator for biologics.
In a recently published study, Coghlan and colleagues designed a semi-automated method for characterizing disulfide bond shuffling on two IgG1 biologics: rituximab (originator drug Rituxan® and biosimilar Acellbia®) and bevacizumab (originator Avastin® and biosimilar Avegra®).
Continue reading “Detecting Disulfide Bond Shuffling in Biologics Using Trypsin Platinum”