In the late-80’s through the 90’s, food and health agencies focused on a mysterious fatal brain disease that infected thousands of cattle. Bovine spongiform encephalitis—or “mad cow disease”—is caused by an infectious protein called a prion. Despite fears that tainted meat would cause the disease to spread to humans, mad cow disease never really made an impact on human health. However, forms of the prion disease such as Creutzfeldt-Jakob disease do affect humans.
In addition to Creutzfeldt-Jakob disease, many neurodegenerative diseases such as Alzheimer’s, Parkinson’s, Huntington’s and amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease) are now thought to be a result of prion-like activity. There is no cure for these diseases, however, new experimental treatment strategies might help slow the progression of neural degeneration.
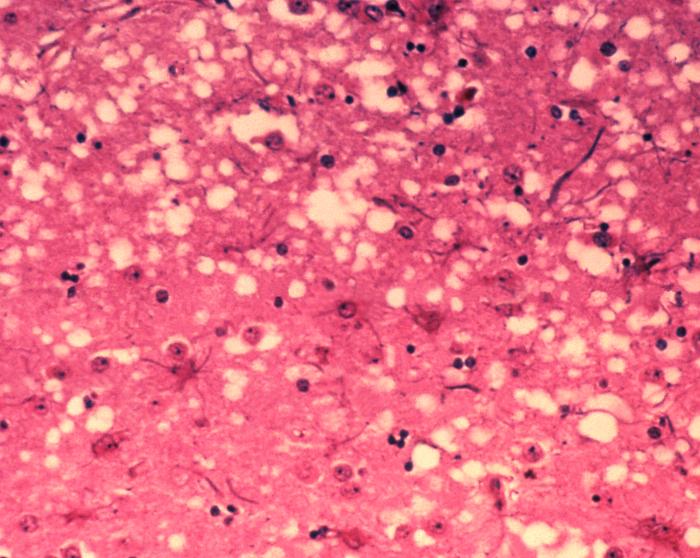
Prions are a misfolded protein in the brain, which create amyloid plaques or “holes” in the brain tissue. Disease progresses as copies of the misfolded protein (PrP) replicate and accumulate. This misfolding results from a mutation in the gene PRNP.
A recent study published by scientists at the National Institute of Allergy and Infectious Diseases/National Institutes of Health (NIAID/NIH) demonstrates that synthetic molecules called antisense oligonucleotides (ASOs) can inhibit the reproduction of the mutated gene and slow the progression of prion disease in mice (1).
Raymond et al. identified two target sequences in the PRNP gene and designed complementary ASOs to mouse PRNP messenger RNA for the 3’ UTR and intron 2 regions of the gene. The researchers tested the ASOs as a prophylactic treatment by injecting mice with two different doses of ASOs into the spinal cord. Then they infected the mice with scrapie, a prion disease that affects sheep and is closely related to Creutzfeldt-Jakob disease in humans. The mice did not show any clinical signs of disease for about 9 months.
They also tested ASOs in mice that were already infected with scrapie. They found that the ASO targeting the 3’ UTR region slowed progression of the disease and the treated mice lived 55% longer than untreated mice.
While animal studies such as this are valuable models for human disease, ASOs are also proving to be successful in human clinical trials. In 2013, Miller et al. conducted the first clinical trial demonstrating that an ASO against the gene SOD1 could be a safe treatment option for patients with ALS (2). Earlier this year at the the American Academy of Neurology’s 71st Annual Meeting, Miller et al. presented preliminary data suggesting that their experimental therapy has the potential to slow ALS disease progression in humans (3).
The new study included 50 patients with SOD1-related ALS. The patients received five injections over 3 months, with either a placebo or varying doses of an ASO drug called tofersen, which targets the mutant SOD1 gene. 10 of the patients who received the highest dose of the drug had a 37% reduction of SOD1 protein in their spinal fluid samples. These findings are pending publication in a peer-reviewed journal.
Time will tell how effective this treatment is at inhibiting progression of the degenerative disease. Additionally, clinical trials are a lengthy process, with a strong need to continue with greater numbers of participants and longer trial periods. But this early evidence suggests that ASOs just might be a successful option to reduce the burden of diseases like ALS and Alzheimer’s in patients hoping to maintain their quality of life.
References:
1) Raymond, GJ et al. (2019) Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight. 4(16):e131175.
2) Miller, TM et al. (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12(5):423.
3) Experimental drug shows promise for genetic form of ALS. [Press release] https://www.aan.com/PressRoom/Home/PressRelease/2719
Latest posts by Mariel Mohns (see all)
- Sustainability Makeover: Parking Ramp Edition - January 27, 2020
- Go with Your Gut: Understanding How the Microbiome and Diet Influence Health - November 26, 2019
- Kyle Hill Explains the Science of Star Wars at the Wisconsin Science Festival - October 23, 2019
Please hold big pharma and the medical community accountable as we should already have a cure or a vaccine/prevention for ALS, Alzheimers, cancer, etc. if we can put people on the moon and in space, we should have already been able to cure or prevent these deadly diseases.