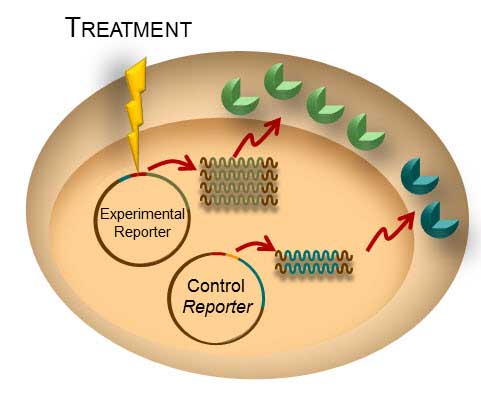
Most, if not all, processes within a cell involve protein-protein interactions, and researchers are always looking for better tools to investigate and monitor these interactions. One such tool is the protein complementation assay (PCA). PCAs use a reporter, like a luciferase or fluorescent protein, separated into two parts (A and B) that form an active reporter (AB) when brought together. Each part of the split reporter is attached to one of a pair of proteins (X and Y) forming X-A and Y-B. If X and Y interact, A and B are brought together to form the active enzyme (AB), creating a luminescent or fluorescent signal that can be measured. The readout from the PCA assay can help identify conditions or factors that drive the interaction together or apart.
A key consideration when splitting a reporter is to find a site that will allow the two parts to reform into an active enzyme, but not be so strongly attracted to each other that they self-associate and cause a signal, even in the absence of interaction between the primary proteins X and Y. This blog will briefly describe how NanoLuc® Luciferase was separated into large and small fragments (LgBiT and SmBiT) that were individually optimized to create the NanoBiT® Assay and show how the design assists in monitoring protein-protein interactions.
Continue reading “For Protein Complementation Assays, Design is Everything”

 One piece of advice you will get from our Technical Services and R&D Scientists with regard to cell-based assays is to pay attention to what you are doing. Sounds obvious, but sloppiness can easily enter into the equation. Do you always count your viable cells with a hemocytometer and trypan blue exclusion before you split a culture? Do you always make sure that each well of your plate or plates contain the same number of cells? Two of our scientists, Terry Riss and Rich Moravec, published a paper demonstrating how decisions you make in experimental setup can ultimately affect the results you obtain. A natural consequence of this is difficulty replicating experiments if you didn’t pay attention to the details during the initial experimental setup.
One piece of advice you will get from our Technical Services and R&D Scientists with regard to cell-based assays is to pay attention to what you are doing. Sounds obvious, but sloppiness can easily enter into the equation. Do you always count your viable cells with a hemocytometer and trypan blue exclusion before you split a culture? Do you always make sure that each well of your plate or plates contain the same number of cells? Two of our scientists, Terry Riss and Rich Moravec, published a paper demonstrating how decisions you make in experimental setup can ultimately affect the results you obtain. A natural consequence of this is difficulty replicating experiments if you didn’t pay attention to the details during the initial experimental setup. 

![4498MA-[Converted]](https://www.promegaconnections.com/wp-content/uploads/2014/10/4498MA-Converted-e1413386878386.jpg)