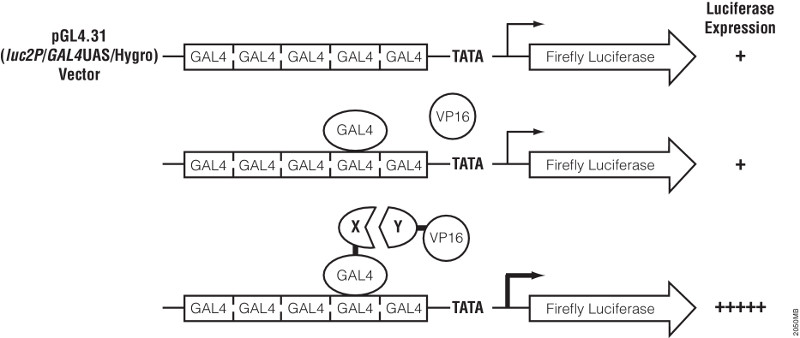
Transcriptional activator-like effector nucleases (TALENs) have rapidly become a technique of choice for precision genome engineering. TALENs are custom-designed nucleases that consist of a modular DNA-binding domain fused to a monomeric, C-terminal FokI nuclease domain (1). TALENs work in pairs and are designed to recognize and bind to tandem-oriented sequences in genomic DNA, separated by a short spacer (15–30 bp). TALEN binding causes dimerization and activation of the FokI nuclease domains, which results in cleavage of the DNA within the spacer region. Small insertions or deletions (indels) are frequently introduced at this site, as the result of errors made during DNA repair by nonhomologous end-joining (NHEJ). These indels can be up to several hundred base pairs in length and result in frameshift mutations that lead to the production of truncated or nonfunctional proteins.
Successful use of TALENs for inducing targeted mutations has been reported in many conventional models, for example: mice, Xenopus and D. melanogaster. TALENs are also reported to be functional in a variety of other invertebrate arthropods, including mosquitos,silkworm and cricket. A recent publication (2) illustrates the use of TALEN technology for the genetic manipulation in P. dumerilii (marine ragworm).
Continue reading “Use of Cell-Free Technology to Evaluate Nuclease (TALEN) Activity on Target DNA”
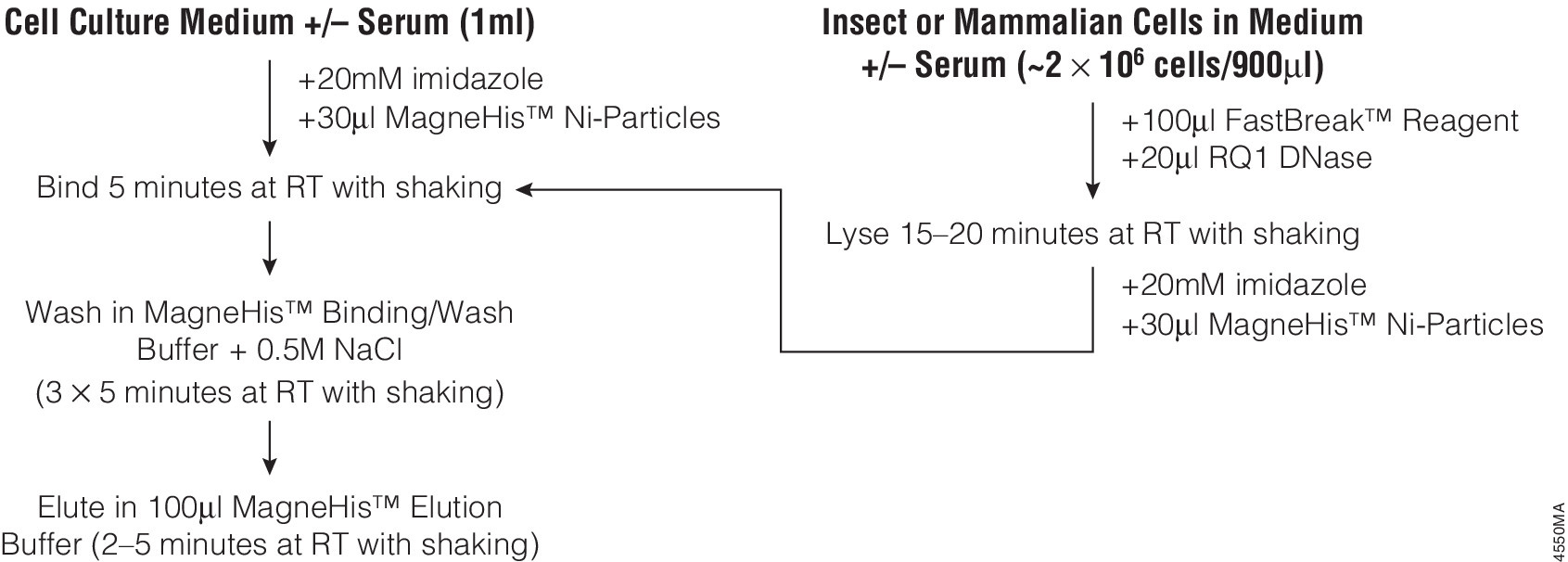
 Many different polypeptide fusion partners or affinity tags have been developed to facilitate purification of target proteins. The most commonly used tag for the purification and detection of recombinant expressed proteins is the His tag. Cloning vectors designed to generate His-tagged proteins contain 5–10 histidine residues at either the C- or N terminus of the expressed protein. The His tag adds only 0.84kDa to the mass of the protein and is nonimmunogenic. Also, because the tertiary structure of the tag is not important for purification, His-tagged proteins can be purified using native or denaturing conditions. The affinity of histidine residues for immobilized nickel allows selective purification of His-tagged proteins. The
Many different polypeptide fusion partners or affinity tags have been developed to facilitate purification of target proteins. The most commonly used tag for the purification and detection of recombinant expressed proteins is the His tag. Cloning vectors designed to generate His-tagged proteins contain 5–10 histidine residues at either the C- or N terminus of the expressed protein. The His tag adds only 0.84kDa to the mass of the protein and is nonimmunogenic. Also, because the tertiary structure of the tag is not important for purification, His-tagged proteins can be purified using native or denaturing conditions. The affinity of histidine residues for immobilized nickel allows selective purification of His-tagged proteins. The