Antibodies labeled with small molecules such as fluorophore, biotin or drugs play a critical role in various areas of biological research,drug discovery and diagnostics. There are several limitations to current methods for labeling antibodies including the need for purified antibodies at high concentrations and multiple buffer exchange steps.
In a recent publication, a method (on-bead conjugation) is described that addresses these limitations by combining antibody purification and conjugation in a single workflow. This method uses high capacity-magnetic Protein A or Protein G beads to capture antibodies directly from cell media followed by conjugation with small molecules and elution of conjugated antibodies from the beads.
Using a variety of fluorophores the researchers show that the on-bead conjugation method is compatible with both thiol- and amine-based chemistry.
This method enables simple and rapid processing of multiple samples in parallel with high-efficiency antibody recovery. It is further shown that recovered antibodies are functional and compatible with downstream applications.
Ricin, derived from caster seeds, inhibits protein synthesis by binding to ribosomes, resulting in cell death. The protein is composed of two polypeptide chains: Ricin Toxin A chain and Ricin Toxin B chain. Ricin inhibits protein synthesis very quickly, and the cell or tissue damage begins within several hours. However, signs of poisoning often are not noted before significant damage has been done, making treatment difficult. Therapeutics that either block the ribosome binding site or compete with the toxin for binding are highly desired. Both antibodies and competitive ligands inhibited binding of the toxin to cell membranes.
A recent publication by Dong et al. (1), described a study to investigate the therapeutic effect of mAb 4C13, a monoclonal antibody against ricin. One of first experiments performed was to determine the general effect the inhibition of protein synthesis induced by ricin using cell-free expression.
In the study, the authors used T3 Coupled Reticulocyte Lysate Systems from Promega. Both ricin and mAb were diluted with saline. Aliquots of ricin (80 ng/ml) were mixed with an equal volume mAbs (1.6μg/ml) or saline alone and incubated at 4 °C for 1.5 h. A total volume of 4μl of sample was added into the reaction system (i.e, T3 Coupled Reticulocyte and plasmid DNA containing the lucifersase gene downstream of T3 RNA phage promoter). After incubation at 30 °C for 1.5 h, the products were cooled at −20 °C for 10 min. A total of 5μl of each reactive product containing synthesized luciferase was mixed with 50μl luciferase assay reagent pre-equilibrated to room temperature, and the fluorescence absorbance was measured immediately with the micro-ELISA Reader.
Positive results obtained from this preliminary experiment, led to more thorough experiments to determine the dosage effect using in vivo models (i.e., cell lines and mice) to characterize the cytotoxicity and binding activity of mAb 4C13. The mAB 4C13 was shown to be a effective in the mouse model.
Protein phosphorylation is a very important protein post-translational modification that controls many cellular processes including metabolism, transcriptional and translation regulation, degradation of proteins, cellular signaling and communication, proliferation, differentiation, and cell survival (1). Approximately 35% of human proteins are phosphorylated. Phosphoproteins are low in abundance, and, therefore, are challenging to detect and characterize by mass spectrometry. Different enrichment systems have been developed to isolate phosphopeptides. Among these techniques, immobilized metal affinity chromatography (IMAC) using Fe3+ and Ga3+ has been widely used for the enrichment of phosphopeptides.
Typical experimental workflows are tedious and consist of numerous steps, including sample collection and cell lysis. One of the major challenges of the process is to maintain the in vivo phosphorylation state of the proteins throughout the preparation process
To evaluate the effect of sample collection protocols on the global phosphorylation status of the cell, a recent paper by Kashin et al. compared different sample workflows by metabolic labeling and quantitative mass spectrometry on Saccharomyces cerevisiae cell cultures (2).
Three different sample collection workflows were evaluated: two that used denaturating conditions and involved mixing of cell cultures with an excess of either ethanol (EtOH) at −80 °C or trichloroacetic acid (TCA), and a third under nondenaturing conditions and washing the cells in PBS.
Their data suggest that either TCA or EtOH sample collection protocols introduced lower collection bias than the PBS protocol. It was also suggested that similar studies be carried out to determine what effects sample preparation has on other post translation modifications such as acetylation or ubiquitination.
Antibody drug conjugates (ADCs) are a new class of therapeutic drugs that uses antibodies to deliver highly toxic drug molecules specifically to the cancer cells. A key requirement for ADCs is the ability of antibody to bind to the cancer cells followed by internalization and subsequent release of drug inside the cells leading to cell apoptosis.
Traditionally, selection of lead antibody candidates for ADCs was done in a sequential workflow where antibodies were first selected based on their affinity followed by characterization involving antibody internalization and drug conjugation. However, there is evidence that high affinity doesn’t always correlate with good internalization and hence there is a need to screen antibodies for internalization properties in addition to their affinities.
Promega has developed a method that allows antibody to be screened for their internalization properties in a simple, plate-based format. The method uses pH sensor dyes (pHAb dyes), which are not fluorescent at neutral pH but become highly fluorescent at acidic pH. When antibody conjugated with pHAb dye binds to its antigen on the cancer cell membrane they are not fluorescent but upon internalization and trafficking into endosomal and lysosomal vesicles the pH drops and dye becomes fluorescent.
Fluorescence signal, for pHAb dyes conjugated using either amine or thiol chemistry, is minimal at pH>7 and increase significantly as the pH drops to pH 5.0, which is a typical pH in cell endosomal compartment. Moreover, pH response of free pHAb dye is similar to that of conjugated dye indicating that conjugation chemistry doesn’t influence the pH response of the dye.
Due to the high signal-to-background ratios of the dyes, plate-based internalization assays can be performed, enabling screening of large libraries of antibodies for their internalization properties, hopefully leading to improved identification of lead candidates for ADC applications.
Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.
Erde et al. (3) described an enhanced FASP (eFASP) workflow that included 0.2% DCA in the exchange, alkylation, and digestion buffers,thus enhancing trypsin proteolysis, resulting in increases cytosolic and membrane protein representation. DCA has been reported (4) to improve the efficiency of the denaturation, solubilization, and tryptic digestion of proteins, particularly proteolytically resistant myoglobin and integral membrane proteins, thereby enhancing the efficiency of their identification with regard to the number of identified proteins and unique peptides.
In a recent publication (5) traditional FASP and eFASP were re-evaluated by ultra-high-performance liquid chromatography coupled to a quadrupole mass filter Orbitrap analyzer (Q Exactive). The results indicate that at the protein level, both methods extracted essentially the same number of hydrophobic transmembrane containing proteins as well as proteins associated with the cytoplasm or the cytoplasmic and outer membranes.
The LC–MS/MS results indicate that FASP and eFASP showed no significant differences at the protein level. However, because of the slight differences in selectivity at the physicochemical level of peptides, these methods can be seen to be somewhat complementary for analyses of complex peptide mixtures.
Crystal Structure of MYC MAX Heterodimer bound to DNA ImageSource=RCSB PDB; StructureID=1nkp; DOI=http://dx.doi.org/10.2210/pdb1nkp/pdb;
In 1982, picked up because of its homology to chicken virus genes that could transform cells, MYC became one of the first human genes identified that could drive cellular transformation (1,2). Since that time countless laboratories have prodded and poked the human MYC gene, the MYC protein, their homologs in other animal models, and their transforming viral counterparts.
MYC is a transcription factor and forms heterodimers with a required protein partner, MAX, before binding to the E box sequences of DNA regulatory regions (3). MYC regulates gene expression of many targets through interactions with a host of proteins, often referred to as the MYC Interactome (2). In fact, MYC is estimated to bind 10–15% of the genome, and it regulates the expression of genes that are transcribed by by each of the three RNA polymerases (2).
MYC plays a central role in regulating cell growth, proliferation, apoptosis, differentiation and transformation, acting as a central integrator of cellular signals. MYC is tightly regulated at multiple levels from gene expression to protein stability. Dysregulation (usually upregulation) of the amount and stability of Myc protein is observed in many human cancers. Even in cancers in which MYC is not directly involved in transforming cells, its normal expression is often required to support the extracellular matrix and/or vascularization necessary for tumor growth and formation (4).
Because MYC is such a central player cancer pathology, it is an attractive target for cancer therapeutics (2) .
Antibodies labelled with radioisotopes or the sequential administrationof an antibody and a radioactive secondary agent facilitate the in vivo detection and/or characterisation of cancers by positron emission tomography (PET) or by single-photon emission computed tomography (SPECT) imaging.
There are drawbacks to both methods, including prolonged exposure to radiation and ensuring that both the antibody and the radiolabelled secondary agent are suitably designed so that they bind rapidly upon contact at the tumor.
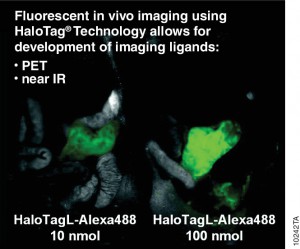
A recent publication (1) investigated a alternative method utilizing the HaloTag® dehalogenase enzyme HaloTag® is a dehalogenase enzyme (33 kDa) that contains an engineered cavity designed to accommodate the reactive chloroalkane group of a HaloTag® ligand (HTL). Upon entering the enzyme cavity, the terminal chlorine atom rapidly undergoes nucleophilic displacement, and a covalent adduct is formed, effectively anchoring the HaloTag® ligand in a precise location.
Three new HaloTag® ligands were synthesized and each labelled with the SPECT radionuclide indium-111 111In-HTL-1 and the dual-modality HaloTag® ligands,111In-HTL-2 and111;In-HTL-3 containing TMR which allows complementary imaging data).
For the validation of the pretargeting strategy based on these HaloTag® ligands, the target human epidermal growth factor receptor 2 (HER2)was selected. Trastuzumab (Herceptin®) was selected as the primary targeting agent and was modified with HaloTag® protein via the trans-cyclooctene/tetrazine ligation.
All three 111In-labelled HaloTa®g ligands exhibited significantly higher binding to the HER2 expressing when compared to negative controls.
Data generated by scientific instruments and decisions based on that data depend on optimal instrument performance. Clinical assays rely on mass spectrometric (MS) data for accurate results so that correct health related results are gained and appropriate results-based decisions are made. However, there are no generally agreed upon tools nor performance standards for mass spectrometry. Furthermore, while several software tools exist that serve to assist with the analysis of instrument performance, a dedicated reagent software package has yet to be created. For optimal liquid chromatography (LC) performance, parameters like retention time, peak width and peak heightare typically reported. Commonly monitored MS parameters include mass accuracy, mass resolution, signal-to noise, sensitivity, limit of detection (LOD), limit of quantitation (LOQ) and dynamic range.
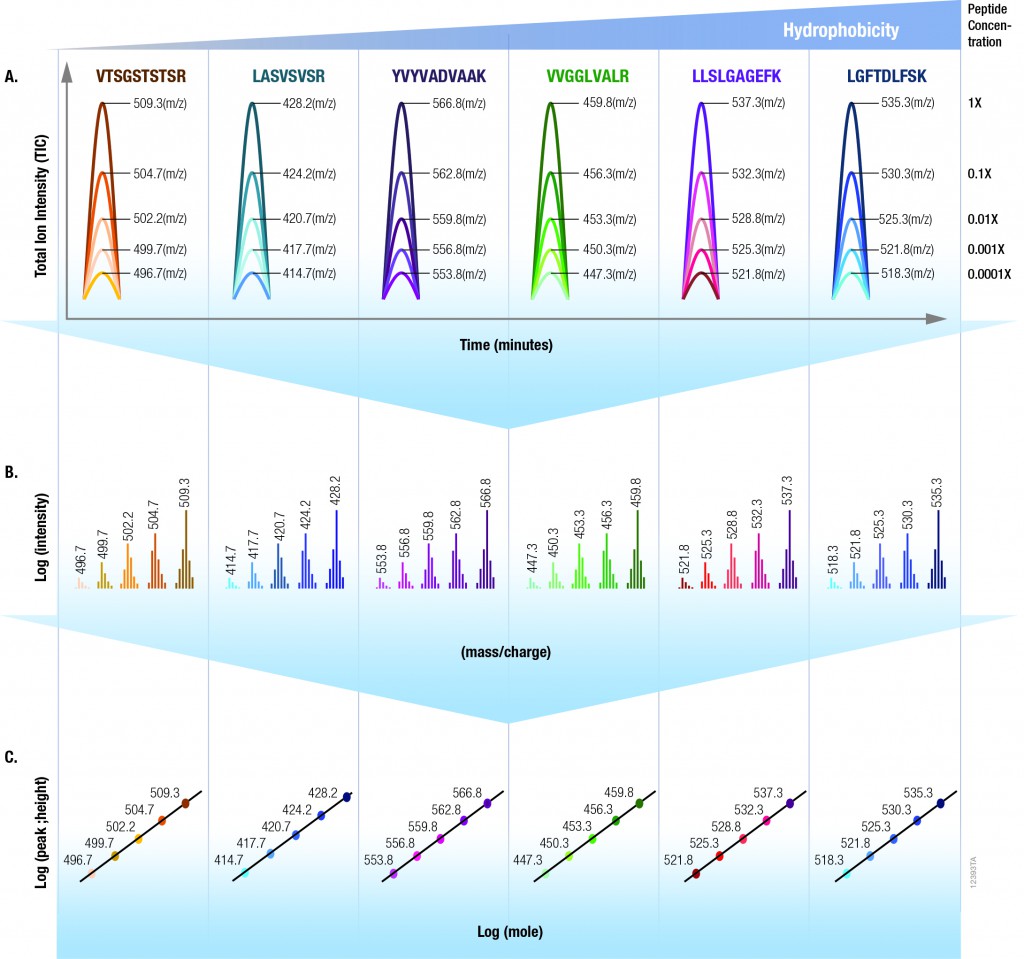
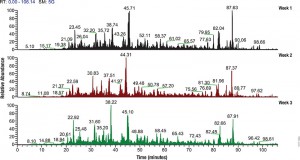
The 6 × 5 LC-MS/MS Peptide Reference Mix is designed for use in method development and optimization,and for routine liquid chromatography (LC) and mass spectrometry (MS) instrument performance monitoring. The product is a mixture of 30 peptides: 6 sets of 5 isotopologues of the same peptide sequence. The isotopologues (Figure 1) differ only by the number of stable, heavy-labeled amino acids incorporated into the sequence. The labeled amino acids consist of uniform 13C and 15N atoms. Each of the isotopologues is indistinguishable chemically and chromatographically. However, since they differ in mass, they are clearly resolved by mass spectrometry.
Figure 1.
The isotopologues of each peptide are present in a series of tenfold differences in concentration or molar abundance. If 1pmol of the mixture is loaded onto an LC column, the next lighter isotopologue would be 100fmol, the next 10fmol, the second lightest 1fmol, and the lightest 100amol. This range allows assessment of the instrument’s dynamic range and sensitivity from a single run.
Peptides with a wide range of hydrophobicities were chosen to enable reporting of LC column performance. The most hydrophilic peptide gives users a tool to optimize the capture of hydrophilic peptides that might be difficult to capture otherwise, but that are too precious to use for method development.
To assist in data processing, a complementary software tool, is provided, the 6 × 5 LC-MS/MS Peptide Reference Mix Analysis Software (The PReMiS™ Software). The PReMiS™ Software produces a tabular report of calculated instrument parameters, graphical analysis of linearity curves as well as reporting the history of user-selected parameters such as LC retention time, peak height and mass accuracy. If the laboratory has a collection of instruments, there is also an option to compare parameters across instruments.
N-Glycosylation is a common protein post-translational modification occurring on asparagine residues of the consensus sequence asparagine-X-serine/threonine, where X may be any amino acid except proline. Protein N-glycosylation takes place in the endoplasmic reticulum (ER) as well as in the Golgi apparatus.
Approximately half of all proteins typically expressed in a cell undergo this modification, which entails the covalent addition of sugar moieties to specific amino acids. There are many potential functions of glycosylation. For instance, physical properties include: folding, trafficking, packing, stabilization and protease protection. N-glycans present at the cell surface are directly involved in cell−cell or cell−protein interactions that trigger various biological responses.
The standard method used to profile the N-glycosylation pattern of cells is glycoprotein isolation followed by denaturation and/or tryptic digestion of the glycoproteins and an enzymatic release of the N-glycans using PNGase F followed by analysis mass spec. This method has been reported to yield high levels of high-mannose N-glycans that stem from both membrane proteins as well as proteins from the ER.(1,2)
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
For those researchers interested in characterizing only cell surface glycans (i.e., complex N-glycans) a recent reference has developed a model system using HEK-292 cells that demonstrates a reproducible, sensitive, and fast method to profile surface N-glycosylation from living cells (3). The method involves standard centrifugation followed by enzymatic release of cell surface N-glycans. When compared to the standard methods the detection and quantification of complex-type N-glycans by increased their relative amount from 14 to 85%.
Protein phosphorylation is the most widespread type of post-translational modification. It affects every basic cellular process, including metabolism, growth, division, differentiation, motility, organelle trafficking, membrane transport, muscle contraction, immunity, learning and memory (1,2). Protein kinases catalyse the transfer of the phosphate from ATP to specific amino acids in proteins. In eukaryotes, these are usually Ser, Thr and Tyr residues. Due to the development of specific phosphopeptide enrichment techniques and highly sensitive MS instruments, phosphoproteomics has enabled researchers to gain a comprehensive view on the dynamics of protein phosphorylation and phosphorylation based signaling networks.
Due to its high cleavage specificity, trypsin is the commonly used proteolytic enzyme in MS-based proteomics, cleaving peptides carboxyterminal of the amino acids lysine and arginine. However, various factors such as the tertiary structure of a protein, adjacent basic amino acids or negatively charged residues close to cleavage sites as well as PTMs are known to impair proteolysis.
To gain closer insights into the impact of phosphorylation on tryptic digestion, a recent publication(3) systematically characterized the digestion efficiency of model peptide sequences that are known to be prone to incomplete digestion.
The results indicated that increasing trypsin concentrations up to a trypsin to peptide ratio of 1:10 led to a significant gain (1) in the overall number of phosphorylation sites (up to 9%) and in the intensities of individual phosphopeptides, thereby improving the sensitivity of phosphopeptide quantification.
The effect of organic solvents (ACN, acetonitrile and TFE trifuorethanol was also evaluated). Positive results were noted with TFE when determining the digestion of individual peptides. However TFE interfered with TiO2 phosphopeptide enrichment and therefore was not recommended for use with complex samples.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.
 Antibodies labeled with small molecules such as fluorophore, biotin or drugs play a critical role in various areas of biological research,drug discovery and diagnostics. There are several limitations to current methods for labeling antibodies including the need for purified antibodies at high concentrations and multiple buffer exchange steps.
Antibodies labeled with small molecules such as fluorophore, biotin or drugs play a critical role in various areas of biological research,drug discovery and diagnostics. There are several limitations to current methods for labeling antibodies including the need for purified antibodies at high concentrations and multiple buffer exchange steps. Ricin, derived from caster seeds, inhibits protein synthesis by binding to ribosomes, resulting in cell death. The protein is composed of two polypeptide chains: Ricin Toxin A chain and Ricin Toxin B chain. Ricin inhibits protein synthesis very quickly, and the cell or tissue damage begins within several hours. However, signs of poisoning often are not noted before significant damage has been done, making treatment difficult. Therapeutics that either block the ribosome binding site or compete with the toxin for binding are highly desired. Both antibodies and competitive ligands inhibited binding of the toxin to cell membranes.
Ricin, derived from caster seeds, inhibits protein synthesis by binding to ribosomes, resulting in cell death. The protein is composed of two polypeptide chains: Ricin Toxin A chain and Ricin Toxin B chain. Ricin inhibits protein synthesis very quickly, and the cell or tissue damage begins within several hours. However, signs of poisoning often are not noted before significant damage has been done, making treatment difficult. Therapeutics that either block the ribosome binding site or compete with the toxin for binding are highly desired. Both antibodies and competitive ligands inhibited binding of the toxin to cell membranes. Protein phosphorylation is a very important protein post-translational modification that controls many cellular processes including metabolism, transcriptional and translation regulation, degradation of proteins, cellular signaling and communication, proliferation, differentiation, and cell survival (1). Approximately 35% of human proteins are phosphorylated. Phosphoproteins are low in abundance, and, therefore, are challenging to detect and characterize by mass spectrometry. Different enrichment systems have been developed to isolate phosphopeptides. Among these techniques, immobilized metal affinity chromatography (IMAC) using Fe3+ and Ga3+ has been widely used for the enrichment of phosphopeptides.
Protein phosphorylation is a very important protein post-translational modification that controls many cellular processes including metabolism, transcriptional and translation regulation, degradation of proteins, cellular signaling and communication, proliferation, differentiation, and cell survival (1). Approximately 35% of human proteins are phosphorylated. Phosphoproteins are low in abundance, and, therefore, are challenging to detect and characterize by mass spectrometry. Different enrichment systems have been developed to isolate phosphopeptides. Among these techniques, immobilized metal affinity chromatography (IMAC) using Fe3+ and Ga3+ has been widely used for the enrichment of phosphopeptides.
 Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.
Filter-aided sample preparation (FASP) method is used for the on-filter digestion of proteins prior to mass-spectrometry-based analyses (1,2). FASP was designed for the removal of detergents, and chaotropes that were used for sample preparation. In addition, FASP removes components such as salts, nucleic acids and lipids. Akylation of reduced cysteine residues is also carried out on filter, after which protein is proteolyzed by use of trypsin on filter in the optimal buffer of the enzyme. Subsequent elution and desalting of the peptide-rich solution then provides a sample ready for LC–MS/MS analysis.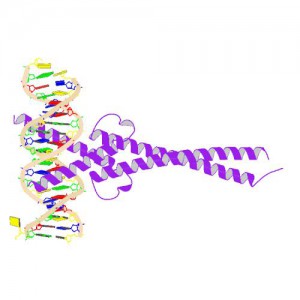
 Antibodies labelled with radioisotopes or the sequential administrationof an antibody and a radioactive secondary agent facilitate the in vivo detection and/or characterisation of cancers by positron emission tomography (PET) or by single-photon emission computed tomography (SPECT) imaging.
Antibodies labelled with radioisotopes or the sequential administrationof an antibody and a radioactive secondary agent facilitate the in vivo detection and/or characterisation of cancers by positron emission tomography (PET) or by single-photon emission computed tomography (SPECT) imaging.