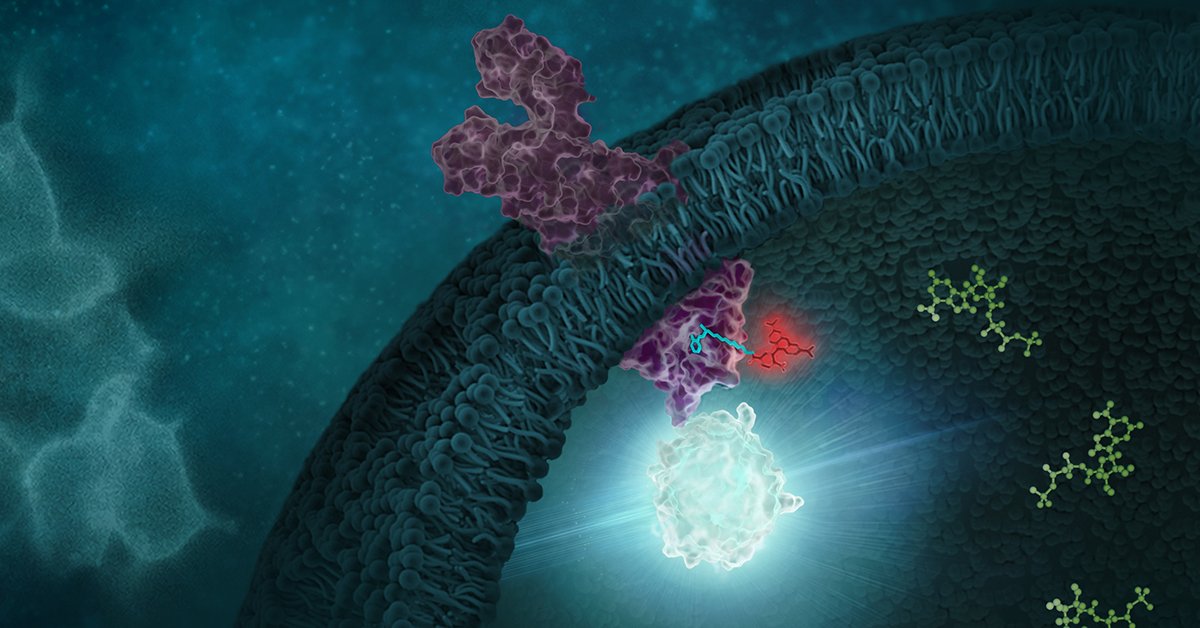
G Protein-Coupled Receptors (GPCRs) are a very large, diverse family of transmembrane receptors in eukaryotes. These receptors detect molecules outside the cell and activate internal signaling pathways by coupling with G proteins. Once a GPCR is activated, β-arrestins translocate to the cell membrane and bind to the occupied receptor, uncoupling it from G proteins and promoting its internalization.
Reporter tags are useful for studying the dynamics of GPCRs and associated proteins, but large tags can disrupt the receptors’ native functioning, and often overexpression of the tagged protein is required to obtain sufficient signal. Here is one example of how researchers have used the small, bright NanoLuc® luciferase to overcome these common challenges and answer questions about GPCRs.
The use of mass spectrometry for the characterization of individual or complex protein samples continues to be one of the fastest growing fields in the life science market.
Bottom-up proteomics is the traditional approach to address these questions. Optimization of each the individual steps (e.g. sample prep, digestion and instrument performance) is critical to the overall success of the entire experiment.
To address issues that may arise in your experimental design, Promega has developed unique tools and complementary webinars to help you along the way.
Here you can find a summary of individual webinars for the following topics:
No protein is an island. Within a cell, protein-protein interactions (PPIs) are involved in highly regulated and specific pathways that control gene expression and cell signaling. The disruption of PPIs can lead to a variety of disease states, including cancer.
Two general approaches are commonly used to study PPIs. Real-time assays measure PPI activity in live cells using fluorescent or luminescent tags. A second approach includes methods that measure a specific PPI “after the fact”; popular examples include a reporter system, such as the classic yeast two-hybrid system.
Dioxins (e.g., 2,3,7,8-Tetrachlorodibenzo-p-dioxin, TCDD) and related compounds (DRCs) are persistent environmental pollutants that gradually accumulate through the food chain, mainly in the fatty tissues of animals. Dioxins are highly toxic and can cause reproductive and developmental problems, damage the immune system, interfere with hormones and also cause cancer. This broad range of toxic and biological effects of DRCs is mostly mediated by the aryl hydrocarbon receptor (AHR).
In animal cells, DRCs bind to AHR in the cytoplasm and then translocate into the nucleus, where they affect the transcription of multiple target genes, including xenobiotic-metabolizing enzymes, such as CYP1A isozymes. AHR is also involved in immune system maintenance, protein degradation and cell proliferation.
The jungle crow (Corvus macrorhynchos) has been considered a suitable indicator for monitoring environmental chemicals such as DRCs. While mammals only have one AHR form, avian species have multiple AHR isoforms such as AHR1 and AHR2. To unveil the functional diversity of multiple avian AHR isoforms in terms of their contribution to responses to DRCs a recent study by Kim et al. investigated the molecular and functional characteristics of jungle crow AHR isoforms, cAHR1 and jcAHR2 (1).
cAHR1 and jcAHR2 proteins were synthesized using AHR proteins were synthesized using the TnT Quick-Coupled Reticulocyte Lysate System to examine whether these jcAHRs have the potential to bind to TCDD. TCDD-binding affinity of the in vitro-expressed jcAHR protein was analyzed using the velocity sedimentation assay with a sucrose gradient.
The results demonstrate that both jcAHR1and jcAHR2 are capable of binding to TCDD.
In a recent reference, Kinoshita and colleagues characterized the phosphorylation dynamics of MEK1 in human cells by using the phosphate affinity electrophoresis technique, Phos-tag sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Phos-tag SDS-PAGE; 1). They found that multiple variants of MEK1 with diferent phosphorylation states are constitutively present in typical human cells.
To investigate the relationships between kinase activity and drug efficacy researchers from the same laboratory group conducted phosphorylation profling of various MEK1 mutants by using Phos-tag SDS- PAGE (2).
Now that Promega is expanding its offerings of options for examining live-cell protein interactions or quantitation at endogenous protein expression levels, we in Technical Services are getting the question about which option is better. The answer is, as with many assays… it depends! First let’s talk about what are the NanoBiT and NanoBRET technologies, and then we will provide some similarities and differences to help you choose the assay that best suits your individual needs.
Large-scale analyses of the proteome have revealed proteomic changes in response to disease, and these changes hold great promise for diagnostics and treatment of complex diseases if proteomic analysis can be brought into the clinical laboratory. Successful and reliable large-scale proteomics requires sample preparation workflows that are reproducible, reliable and show little variability. To bring proteomics into the clinical laboratory, standardized procedures and workflows for sample prep and analysis are required to generate valid, actionable results on a time scale useful for the clinic.
The two most common sample types analyzed for clinical proteomics are body fluids and tissue biopsies. To process these kinds of samples, there are two initial steps: tissue solubilization, followed by proteolytic digestion. Solubilization of solid tissues is the most labor-intensive and produces the most variable results.
The introduction of pressure cycling technology (PCT) using Barocycler instrumentation has greatly improved both tissue solubilization and digestion consistency. The PCT-based sample preparation protocols generally utilize urea as a lysis buffer for protein denaturing and solubilization. Urea has several drawbacks including inhibiting trypsin activity and introducing unwanted modifications like carbamylation.
Lucas and colleagues analyzed whether replacing urea with SDC would produce similar tissue digestion profiles and improve the PCT method.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
SDC allowed the use of higher temperatures compared to urea, and hence the first step (lysis, reduction, and alkylation) was performed at 56 °C. The second digestion step in the Barocycler was optimized, and the third step was eliminated. To further reduce digestion time, they capitalized on Rapid Trypsin/Lys-C. Rapid Trypsin/Lys-C maintains robust activity at 70 °C, and allowed Barocycler digestion to be performed in a single step, completing digestion in 30 cycles (approximately 30 min) rather than 105 minutes, streamlining the protocol.
The data presented an improved conventional tissue PCT approach in a Barocycler by replacing urea and proteolytic enzymes with SDC, N-propanol, and modified commercially available enzymes that have higher optimum temperatures.
Try a sample of high-efficiency Trypsin Platinum today!
Visit our website for more on Trypsin Platinum, Mass Spectrometry Grade, with enhanced proteolytic efficiency and superior autoproteolytic resistance.
Long noncoding RNAs have been shown to regulate chromatin states, transcriptional activity and post transcriptional activity (1). Only a few studies have observed long non-coding RNAs modulating the translational process (2). The noncoding RNA BC200 has been shown to inhibit translation by interacting with the translation initiation factors, eIF4A and eIF4B.
To characterize how BC200 translational inhibition could be controlled, a variety of RNAs were transcribed/translated in vitro using the TNT system (Cat. #L4610) from Promega. To each transcription/translation reaction, BC900 RNA, hnRNPE1 and hnRNE2 proteins were added. Inhibition of BC200 activity was noted when proteins were successful expressed (3).
Literature Cited
Sosinska, P et.al. (2015) Intraperitoneal invasiveness of ovarian cancer from the cellular and molecular perspective. Ginekol. Pol. 86, 782–86.
With the use of a suite of “-omics” technologies you can examine the way in which complex cellular processes work together across all molecular domains (i.e., proteomics, metabolomics, transcriptomics) in a single biological system. Several studies have been published across a wide range of fields illustrating the power of such a unified approach (1,2). Most studies however did not focus on the development of a high-throughput, unified sample preparation approach to complement high-throughput “omic” analytics.
A recent publication by Gutierrez and colleagues presents a simple high-throughput process (SPOT) that has been optimized to provide high-quality specimens for metabolomics, proteomics, and transcriptomics from a common cell culture sample (3). They demonstrate that this approach can process 16−24 samples from a cell pellet to a desalted sample ready for mass spectrometry analysis within 9 hours. They also demonstrated that the combined process did not sacrifice the quality of data when compared to individual sample preparation methods.
It’s time to analyze your protein and you are trying to decide where to begin. You are asking questions like: Which protease do I choose? How much enzyme should I use in my digest? How long should I perform my digest?
Unfortunately, there is no one-size fits all answer to this type of question other than… “well it depends.” All protease digests will be a balance between denaturing the protein sample to allow access to cleavage sites, optimizing conditions for the protease to function, and compatibility with your workflow and downstream applications. We provide general guidelines that work for most samples, but frequently you will need to optimize the conditions need for your specific sample and application.
Here, I use the example of a trypsin digest for downstream mass spectrometry to highlight key questions to ask and factors that can be optimized for any digest.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.