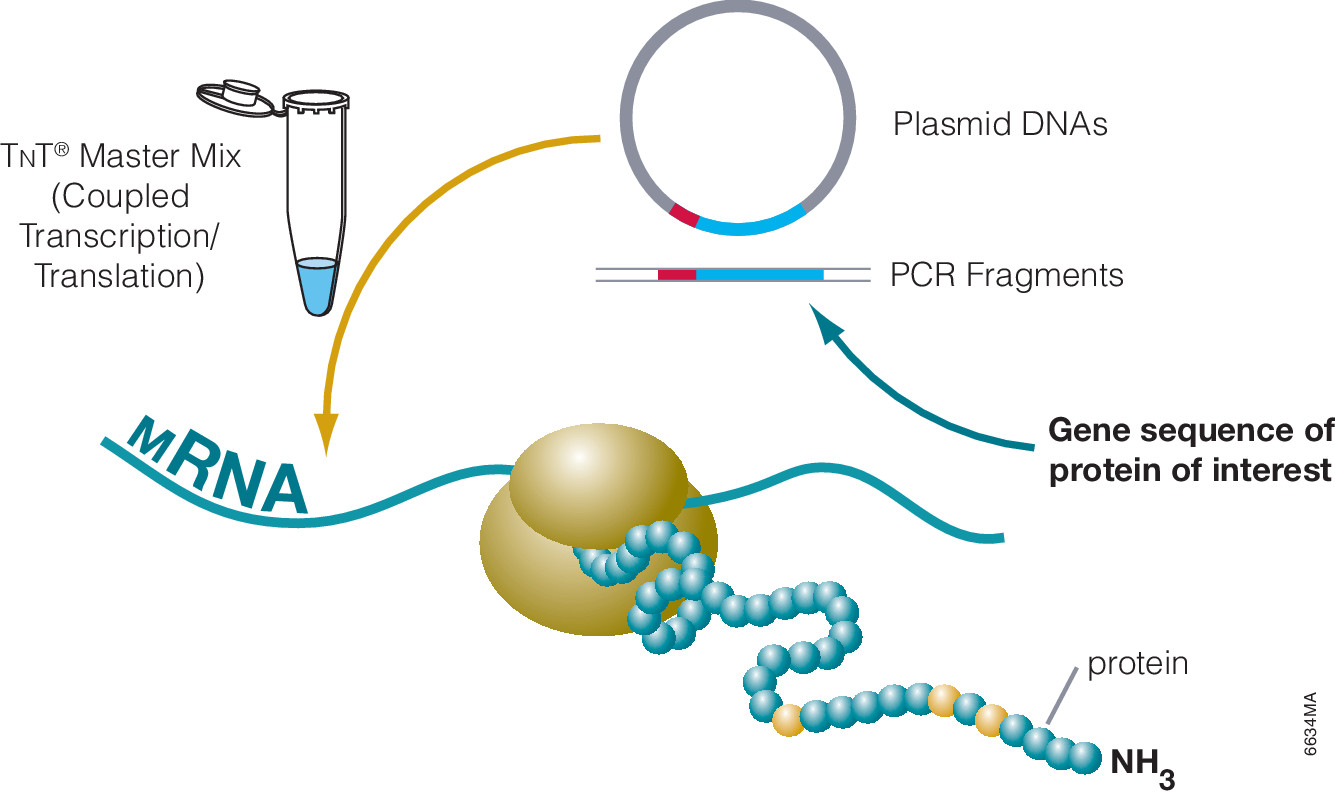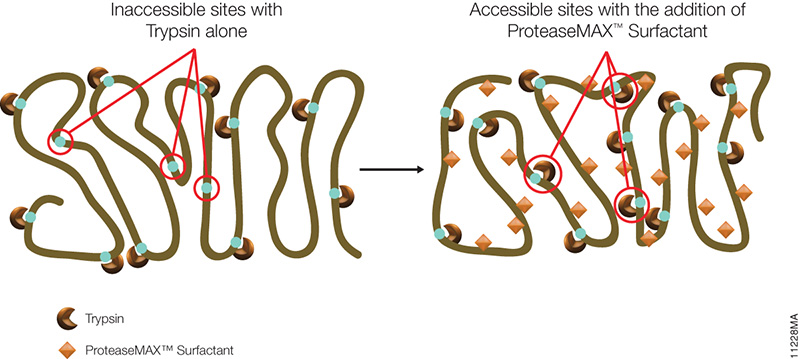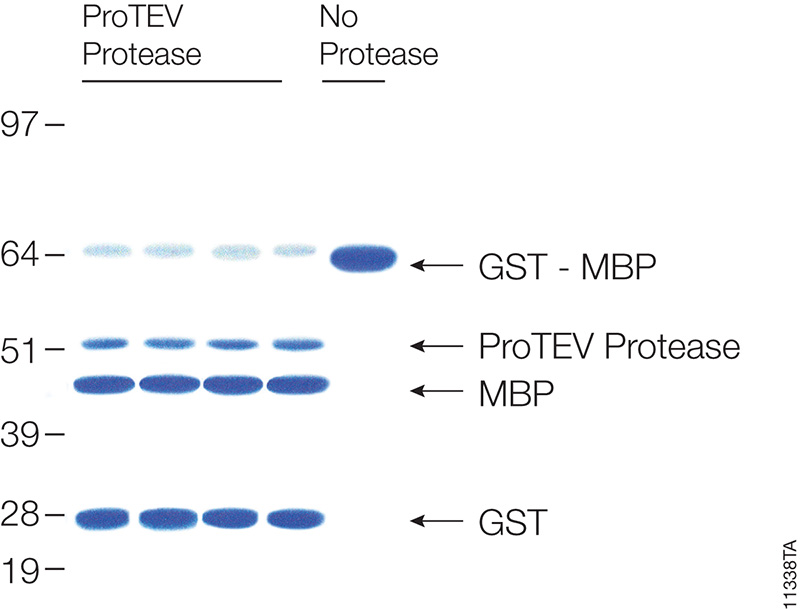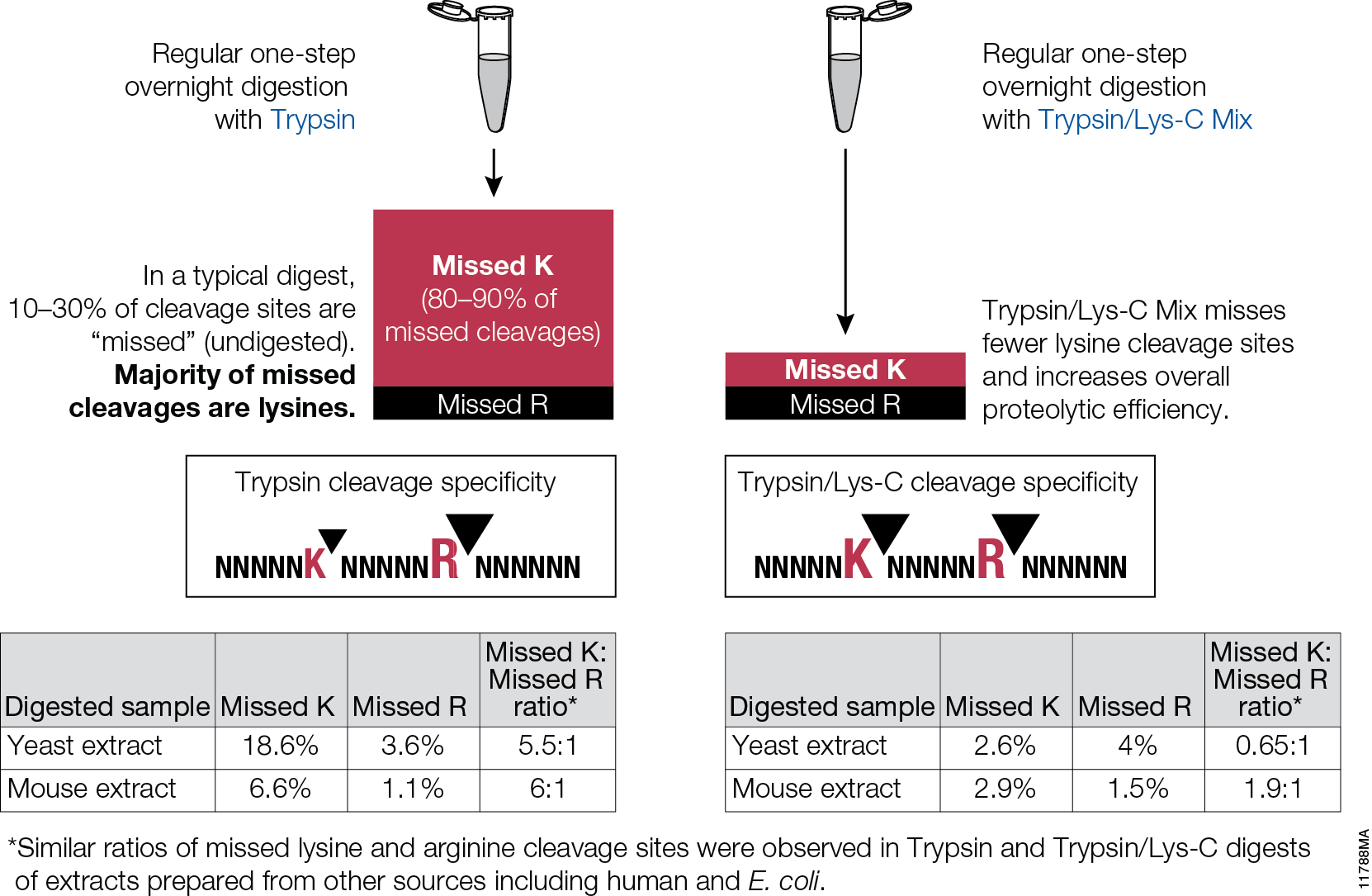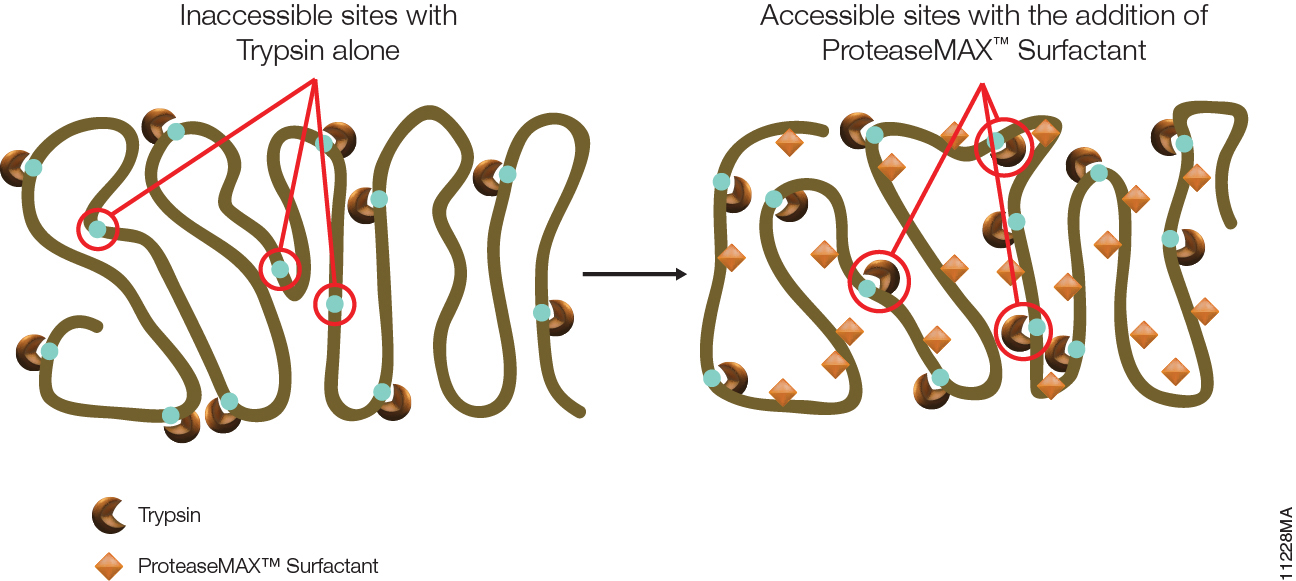
Asp-N, Sequencing Grade, is an endoproteinase that hydrolyzes peptide bonds on the N-terminal side of aspartic and cysteic acid residues: Asp and Cys. Asp-N activity is optimal in the pH range of 4.0–9.0. This sequencing grade enzyme can be used alone or in combination with trypsin or other proteases to produce protein digests for peptide mapping applications or protein identification by peptide mass fingerprinting or MS/MS spectral matching. It is suitable for in-solution or in-gel digestion reactions.
The following references illustrate the use of Asp-N in recent publications:
Protein sequence coverage
- Jakobsson, M et al. (2013) Identification and characterization of a novel Human Methyltransferase modulating Hsp70 protein function through lysine methylation. J. Biol. Chem. 288, 27752–63.
- Carroll, J. et. al. (2013) Post-translational modifications near the quinone binding site of mammalian complex I. J. Biol. Chem. 288, 24799–08.
Glycoprotein analysis
- Siguier, B. et al. (2014) First structural insights into α-L-Arabinofuranosidases from the two GH62 Glycoside hydrolase subfamilies. J. Biol. Chem. 289, 5261–73.
- Vakhrushev, S. et al. (2013) Enhanced mass spectrometric mapping of the human GalNAc-type O-glycoproteome with SimpleCells. Mol. Cell. Prot. 12, 932–44.
- Berk, J. et al. (2013) . O-Linked β-N- Acetylglucosamine (O-GlcNAc) Regulates emerin binding to autointegration Factor (BAF) in a chromatin and Lamin B-enriched “Niche”. J. Biol. Chem. 288, 30192–09.
Phosphoprotein analysis
- Roux, P. and Thibault, P. (2013) The Coming of Age of phosphoproteomics –from Large Data sets to Inference of protein Functions. Mol. Cell. Prot. 12, 3453–64.
Are you looking for proteases to use in your research?
Explore our portfolio of proteases today.
 Although previous references have provided data regarding the potential oncogenic role of the gene ETV7, there has been minimal investigation as to its physiological role.
Although previous references have provided data regarding the potential oncogenic role of the gene ETV7, there has been minimal investigation as to its physiological role.