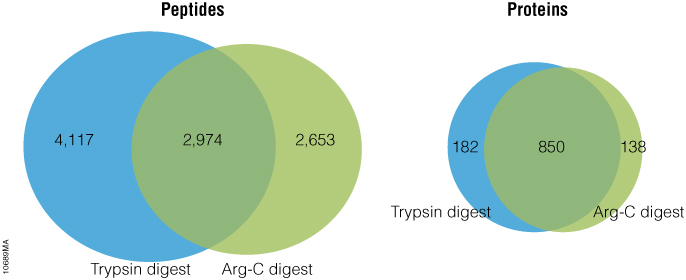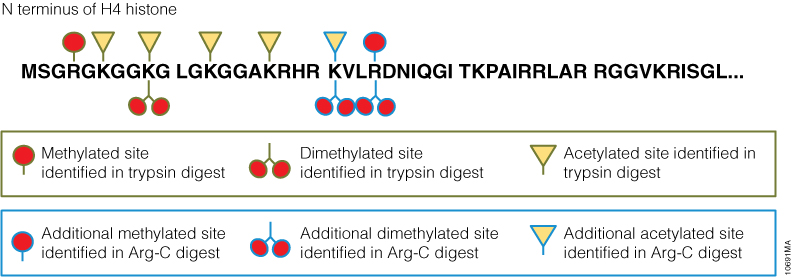
Gel electrophoresis and gel staining are common lab tasks that you may not think too much about. It’s a fairly routine part of your day…purify DNA or RNA, check it on a gel. As you probably know, interchelating agents like ethidium bromide can be used to visualize your nucleic acids on a gel for relatively low cost. The problem with ethidium bromide is that it’s highly mutagenic, making it less than ideal to work with and disposal of ethidium bromide can be quite costly. There are other commercially available alternatives to ethidium bromide that use fluorescent-based dyes to detect nucleic acids in gels. Some of these are touted to be safer than ethidium bromide; others are marketed as more sensitive. If you are going to switch from an interchelating agent to something safer, you certainly don’t want to lose out on sensitivity.
To make your gel staining safer, more convenient, and more cost-effective, we’ve developed the Diamond™ Nucleic Acid Dye. This dye is not detectably genotoxic or cytotoxic at the 1:10,000 dilution recommended for gel staining, as determined by the Ames MPF™ Assay, is more sensitive than competing fluorescent-type “safe” dyes, and, in its concentrated form, is room-temperature stable for 90 days (1, 2). If you are looking to switch to a safer, more sensitive way to stain your polyacrylamide or agarose gels to visualize your DNA or RNA, you may want to give the Diamond™ Nucleic Acid Dye a try.
- Schagat, T. and Hendricksen, A. Diamond™ Nucleic Acid Dye is a Safe and Economical Alternative to Ethidium Bromide. [Internet] July 2013; tpub 125. [cited: 2013, July, 29].
- Truman, A., Hook, B. and Hendricksen, A. Diamond™ Nucleic Acid Dye: A Sensitive Alternative to SYBR® Dyes. [Internet] June 2013; tpub 121. [cited: 2013, July, 29].