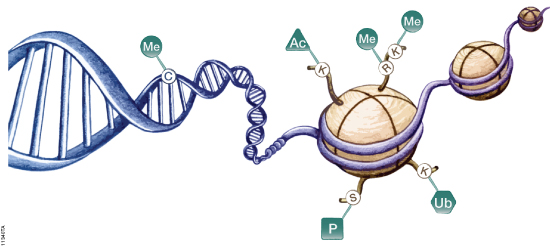
DNA is organized by protein:DNA complexes called nucleosomes in eukaryotes. Nucleosomes are composed of 147 base pairs of DNA wrapped around a histone octamer containing two copies of each core histone protein. Histone proteins play significant roles in many nuclear processes including transcription, DNA damage repair and heterochromatin formation. Histone proteins are extensively and dynamically post-translationally modified, and these post-translational modifications (PTMs) are thought to comprise a specific combinatorial PTM profile of a histone that dictates its specific function. Abnormal regulations of PTM may lead to developmental disorders and disease development such as cancer.
Recombinant erythropoietin (rhEPO) is often used as “doping agent” by athletes in endurance sports to increase blood oxygen capacity. Some strategies improve the pharmacological properties of erythropoietin (EPO) through the genetic and chemical modification of the native EPO protein. The EPO-Fcs are fusion proteins composed of monomeric or dimeric recombinant EPO and the dimeric Fc region of human IgG molecules. The Fc region includes the hinge region and the CH2 and CH3 domains. Recombinant human EPOs (rhEPO) fused to the IgG Fc domain demonstrate a prolonged half-life and enhanced erythropoietic activity in vivo compared with native or rhEPO.
Drug-testing agencies will need to obtain primary structure information and develop a reliable analytical method for the determination of EPO-Fc abuse in sport. The possibility of EPO-Fc detection using nanohigh-performance liquid chromatography−tandem mass spectrometry (HPLC−MS/MS) was already demonstrated (1). However, the prototyping peptides derived from EPO and IgG are not selective enough because both free proteins are naturally presented in human serum. In a recent publication, researchers describe the effort to identify peptides covering unknown fusion breakpoints (later referred to as “spacer” peptides; 2). The identification of “spacer” peptides will allow the confirmation of the presence of exogenous EPO-Fc in human biological fluids.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
A bottom-up approach and the intact molecular weight measurement of deglycosylated protein and its IdeS proteolytic fractions was used to determine the amino acid sequence of EPO-Fc. Using multiple proteases, peptides covering unknown fusion breakpoints (spacer peptides) were identified.
Results indicated that “spacer peptides” could be used in the determination of EPO-Fc fusion proteins in biological samples using common LC−tandem MS methods.
Illustration showing NanoLuc and firefly luciferase reporters.
The luciferase immunoprecipitation system (LIPS) assay is a liquid phase immunoassay allowing high-throughput serological screening of antigen-specific antibodies. The immunoassay involves quantitating serum antibodies by measuring luminescence emitted by the reporter enzyme Renilla luciferase (Rluc) fused to an antigen of interest. The Rluc-antigen fusion protein is recognized by antigen-specific antibodies, and antigen-antibody complexes are captured by protein A/G beads that recognize the Fc region of the IgG antibody (1).
Monoclonal antibodies (mAbs) have been widely used to eliminate undesired cells via various mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and programmed cell death (PCD). Unlike the Fc-dependent mechanism of ADCC and CDC, certain antibody–antigen interactions can evoke direct PCD via apoptosis or oncosis. Previously, researchers have reported the specific killing of undifferentiated human embryonic stem cells (hESC) by mAb84 (IgM) via oncosis (1)
In a recent publication (2), a monoclonal antibody (mAb), TAG-A1 (A1), was generated to selectively kill residual undifferentiated human embryonic stem cells (hESC). One of the many experimental tools used to characterize the mechanism of oncosis was the fragmention of the A1 antibody with IdeS and papain.
Papain digestion of IgG produces Fab fragments in the presence of reducing agent. F(ab)2 fragments of A1 were produced using IdeS Protease.
The results indicate that both Fab_A1 and F(ab)2_A1 bind to hESC but only F(ab)2_A1 retained hESC killing. Hence bivalency, but not Fc-domain, is essential for A1 killing on hESC.
Choo, A.B. et al. (2008) Selection against undifferentiated human embryonic stem cells by a cytotoxic antibody recognizing podocalyxin-like protein-1. Stem Cells26, 1454.
Zheng, J.Y. et al. (2017) Excess reactive oxygen species production mediates monoclonal antibody-induced human embryonic stem cell death via oncosis. Cell Death and Differentiation24, 546–58.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
Several pharmaceutical companies have biosimilar versions of therapeutic mAbs in development. Biosimilars can promise significant cost savings for patients, but the unavoidable differences between the original and thencopycat biologic raise questions regarding product interchangeability. Both innovator mAbs and biosimilars are heterogeneous populations of variants characterized by differences in glycosylation,oxidation, deamidation, glycation, and aggregation state. Their heterogeneity could potentially affect target protein binding through the F´ab domain, receptor binding through the Fc domain, and protein aggregation.
As more biosimilar mAbs gain regulatory approval, having clear framework for a rapid characterization of innovator and biosimilar products to identify clinically relevant differences is important. A recent reference (1) applied a comprehensive mass spectrometry (MS)-based strategy using bottom-up, middle-down, and intact strategies. These data were then integrated with ion mobility mass spectrometry (IM-MS) and collision-induced unfolding (CIU) analyses, as well as data from select biophysical techniques and receptor binding assays to comprehensively evaluate biosimilarity between Remicade and Remsima.
The authors observed that the levels of oxidation, deamidation, and mutation of individual amino acids were remarkably similar. they found different levels of C-terminal truncation, soluble protein aggregates, and glycation that all likely have a limited clinical impact. Importantly, they identified more than 25 glycoforms for each product and observed glycoform population differences.
Overall the use of mass spectrometry-based analysis provides rapid and robust analytical information vital for biosimilar development. They demonstrated the utility of our multiple-attribute monitoring workflow using the model mAbs Remicade and Remsima and have provided a template for analysis of future mAb biosimilars.
While many proteases are used in bottom-up mass spectrometric (MS) analysis, trypsin (4,5) is the de facto protease of choice for most applications. There are several reasons for this: Trypsin is highly efficient, active, and specific. Tryptic peptides produced after proteolysis are ideally suited, in terms of both size (350–1,600 Daltons) and charge (+2 to +4), for MS analysis. One significant drawback to trypsin digestion is the long sample preparation times, which typically range from 4 hours to overnight for most protocols. Achieving efficient digestion usually requires that protein substrates first be unfolded either with surfactants or denaturants such as urea or guanidine. These chemical additives can have negative effects, including protein modification, inhibition of trypsin or incompatibility with downstream LC-MS/MS. Accordingly, additional steps are typically required to remove these compounds prior to analysis.
Researchers and clinicians are fairly certain that all cervical cancers are caused by Human Papillomavirus (HPV) infections, and that HPV16 and HPV18 are responsible for about 70% of all cases. HPV16 and HPV18 have also been shown to cause almost half the vaginal, vulvar, and penile cancers, while about 85% of anal cancers are also caused by HPV16.
E6 is a potent oncogene of HR-HPVs, and its role in progression to malignancy continues to be explored. The E6 oncoprotein of HPV can promote viral DNA replication through several pathways. It forms a complex with human E3-ubiquitin ligase E6-associated protein (E6AP), which can in turn target the p53 tumor-suppressor protein, leading to its ubiquitin-mediated degradation. In particular, E6 from HR-HPVs can block apoptosis, activate telomerase, disrupt cell adhesion, polarity and epithelial differentiation, alter transcription and G-protein signaling, and reduce immune recognition of HPV-infected cells.
Therapeutic monoclonal antibodies (MAbs) are inherently heterogeneous due to a wide range of both enzymatic and chemical modifications, such as oxidation, deamidation and glycosylation which may occur during expression, purification or storage. For identification and functional evaluation of these modifications, stability studies are typically performed by employing stress conditions such as exposure to chemical oxidizers, elevated pH and temperature.
To characterize MAbs, a variety of analytical techniques are chosen, such as size exclusion chromatography and ion exchange chromatography. However, due to the large size of the intact MAbs, these methods lack structural resolution. Often, the chromatographic peaks resolved by SEC and IEC methods are collected and further analyzed by peptide mapping to obtain more detailed information. Peptide mapping, in which antibodies are cleaved into small peptides through protease digestion followed by LC–MS/MS analysis, is generally the method of choice for detection and quantitation of site-specific modifications. However sample preparation and lengthy chromatographic separation make peptide mapping impractical for the analysis of large numbers of samples. In contrast to peptide mapping analysis, the middle-down approach offers the advantage of high-throughput and specificity for antibody characterization.
Limited proteolysis of IgG molecules by the IdeS enzyme has been introduced for antibody characterization due to its high cleavage specificity and simple digestion procedure.
Protein-DNA interactions are fundamental processes in gene regulation in a living cells. These interactions affect a wide variety of cellular processes including DNA replication, repair, and recombination. In vivo methods such as chromatin immunoprecipitation (1) and in vitro electrophoretic mobility shift assays (2) have been used for several years in the characterization of protein-DNA interactions. However, these methods lack the throughput required for answering genome-wide questions and do not measure absolute binding affinities. To address these issues a recent publication (3) presented a high-throughput micro fluidic platform for Quantitative Protein Interaction with DNA (QPID). QPID is an microfluidic-based assay that cam perform up to 4096 parallel measurements on a single device.
The basic elements of each experiment includes oligonucleotides that were synthesized and hybridized to a Cy5-labeled primer and extended using Klenow. All transcription factors that were evaluated contained a 3’HIS and 5’ cMyc tag and were expressed in rabbit reticulocyte coupled transcription and translation reaction (TNT® Coupled Reticulocyte Lysate). Expressed proteins are loaded onto to the QIPD device and immobilized. In the DNA binding assay the fluorescent DNA oligonucleotides are incubated with the immobilized transcription factors and fluorescent images taken. To validate this concept the binding of four different transcription factor complexes to 32 oligonucleotides at 32 different concentrations was characterized in a single experiment. In a second application, the binding of ATF1 and ATF3 to 128 different DNA sequences at different concentrations were analyzed on a single device.
Bottom-up proteomics focuses on the analysis of protein mixtures after enzymatic digestion of the proteins into peptides. The resulting complex mixture of peptides is analyzed by reverse-phase liquid chromatography (RP-LC) coupled to tandem mass spectrometry (MS/MS). Identification of peptides and subsequently proteins is completed by matching peptide fragment ion spectra to theoretical spectra generated from protein databases.
Trypsin has become the gold standard for protein digestion to peptides for shotgun proteomics. Trypsin is a serine protease. It cleaves proteins into peptides with an average size of 700-1500 daltons, which is in the ideal range for MS (1). It is highly specific, cutting at the carboxyl side of arginine and lysine residues. The C-terminal arginine and lysine peptides are charged, making them detectable by MS. Trypsin is highly active and tolerant of many additives.
Even with these technical features, the use of trypsin in bottom-up proteomics may impose certain limits in the ability to grasp the full proteome, Tightly-folded proteins can resist trypsin digestion. Post-translational modifications (PTMs) present a different challenge for trypsin because glycans often limit trypsin access to cleavage sites, and acetylation makes lysine and arginine residues resistant to trypsin digestion.
Are you looking for proteases to use in your research? Explore our portfolio of proteases today.
To overcome these problems, the proteomics community has begun to explore alternative proteases to complement trypsin. However, protocols, as well as expected results generated when using these alternative proteases have not been systematically documented.
In a recent reference (2), optimized protocols for six alternative proteases that have already shown promise in their applicability in proteomics, namely chymotrypsin, Lys-C, Lys-N, Asp-N, Glu-C and Arg-C have been created.
Data describe the appropriate MS data analysis methods and the anticipated results in the case of the analysis of a single protein (BSA) and a more complex cellular lysate (Escherichia coli). The digestion protocol presented here is convenient and robust and can be completed in approximately in 2 days.
Try a sample of high-efficiency Trypsin Platinum today!
Visit our website for more on Trypsin Platinum, Mass Spectrometry Grade, with enhanced proteolytic efficiency and superior autoproteolytic resistance.
XWe use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To learn more about our approach to Privacy we invite you to Read More
By clicking “Accept All”, you consent to the use of ALL the cookies. However you may visit Cookie Settings to provide a controlled consent.
We use cookies and similar technologies to make our website work, run analytics, improve our website, and show you personalized content and advertising. Some of these cookies are essential for our website to work. For others, we won’t set them unless you accept them. To find out more about cookies and how to manage cookies, read our Cookie Policy.
If you are located in the EEA, the United Kingdom, or Switzerland, you can change your settings at any time by clicking Manage Cookie Consent in the footer of our website.
Necessary cookies are absolutely essential for the website to function properly. These cookies ensure basic functionalities and security features of the website, anonymously.
Cookie
Duration
Description
cookielawinfo-checbox-analytics
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Analytics".
cookielawinfo-checbox-functional
11 months
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Functional".
cookielawinfo-checbox-others
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Other.
cookielawinfo-checkbox-advertisement
1 year
The cookie is set by GDPR cookie consent to record the user consent for the cookies in the category "Advertisement".
cookielawinfo-checkbox-necessary
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookies is used to store the user consent for the cookies in the category "Necessary".
cookielawinfo-checkbox-performance
11 months
This cookie is set by GDPR Cookie Consent plugin. The cookie is used to store the user consent for the cookies in the category "Performance".
gdpr_status
6 months 2 days
This cookie is set by the provider Media.net. This cookie is used to check the status whether the user has accepted the cookie consent box. It also helps in not showing the cookie consent box upon re-entry to the website.
lang
This cookie is used to store the language preferences of a user to serve up content in that stored language the next time user visit the website.
viewed_cookie_policy
11 months
The cookie is set by the GDPR Cookie Consent plugin and is used to store whether or not user has consented to the use of cookies. It does not store any personal data.
Analytical cookies are used to understand how visitors interact with the website. These cookies help provide information on metrics the number of visitors, bounce rate, traffic source, etc.
Cookie
Duration
Description
SC_ANALYTICS_GLOBAL_COOKIE
10 years
This cookie is associated with Sitecore content and personalization. This cookie is used to identify the repeat visit from a single user. Sitecore will send a persistent session cookie to the web client.
vuid
2 years
This domain of this cookie is owned by Vimeo. This cookie is used by vimeo to collect tracking information. It sets a unique ID to embed videos to the website.
WMF-Last-Access
1 month 18 hours 24 minutes
This cookie is used to calculate unique devices accessing the website.
_ga
2 years
This cookie is installed by Google Analytics. The cookie is used to calculate visitor, session, campaign data and keep track of site usage for the site's analytics report. The cookies store information anonymously and assign a randomly generated number to identify unique visitors.
_gid
1 day
This cookie is installed by Google Analytics. The cookie is used to store information of how visitors use a website and helps in creating an analytics report of how the website is doing. The data collected including the number visitors, the source where they have come from, and the pages visted in an anonymous form.
Advertisement cookies are used to provide visitors with relevant ads and marketing campaigns. These cookies track visitors across websites and collect information to provide customized ads.
Cookie
Duration
Description
IDE
1 year 24 days
Used by Google DoubleClick and stores information about how the user uses the website and any other advertisement before visiting the website. This is used to present users with ads that are relevant to them according to the user profile.
test_cookie
15 minutes
This cookie is set by doubleclick.net. The purpose of the cookie is to determine if the user's browser supports cookies.
VISITOR_INFO1_LIVE
5 months 27 days
This cookie is set by Youtube. Used to track the information of the embedded YouTube videos on a website.
Performance cookies are used to understand and analyze the key performance indexes of the website which helps in delivering a better user experience for the visitors.
Cookie
Duration
Description
YSC
session
This cookies is set by Youtube and is used to track the views of embedded videos.
_gat_UA-62336821-1
1 minute
This is a pattern type cookie set by Google Analytics, where the pattern element on the name contains the unique identity number of the account or website it relates to. It appears to be a variation of the _gat cookie which is used to limit the amount of data recorded by Google on high traffic volume websites.